DiffFit: Visually-Guided Differentiable Fitting of Molecule Structures to a Cryo-EM Map
Deng Luo - King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
Zainab Alsuwaykit - King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
Dawar Khan - King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
Ondřej Strnad - King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
Tobias Isenberg - Université Paris-Saclay, CNRS, Orsay, France. Inria, Saclay, France
Ivan Viola - King Abdullah University of Science and Technology, Thuwal, Saudi Arabia
Download preprint PDF
Download Supplemental Material
Room: Bayshore I
2024-10-16T14:15:00ZGMT-0600Change your timezone on the schedule page
2024-10-16T14:15:00Z
Fast forward
Full Video
Keywords
Scalar field data, algorithms, application-motivated visualization, process/workflow design, life sciences, health, medicine, biology, structural biology, bioinformatics, genomics, cryo-EM
Abstract
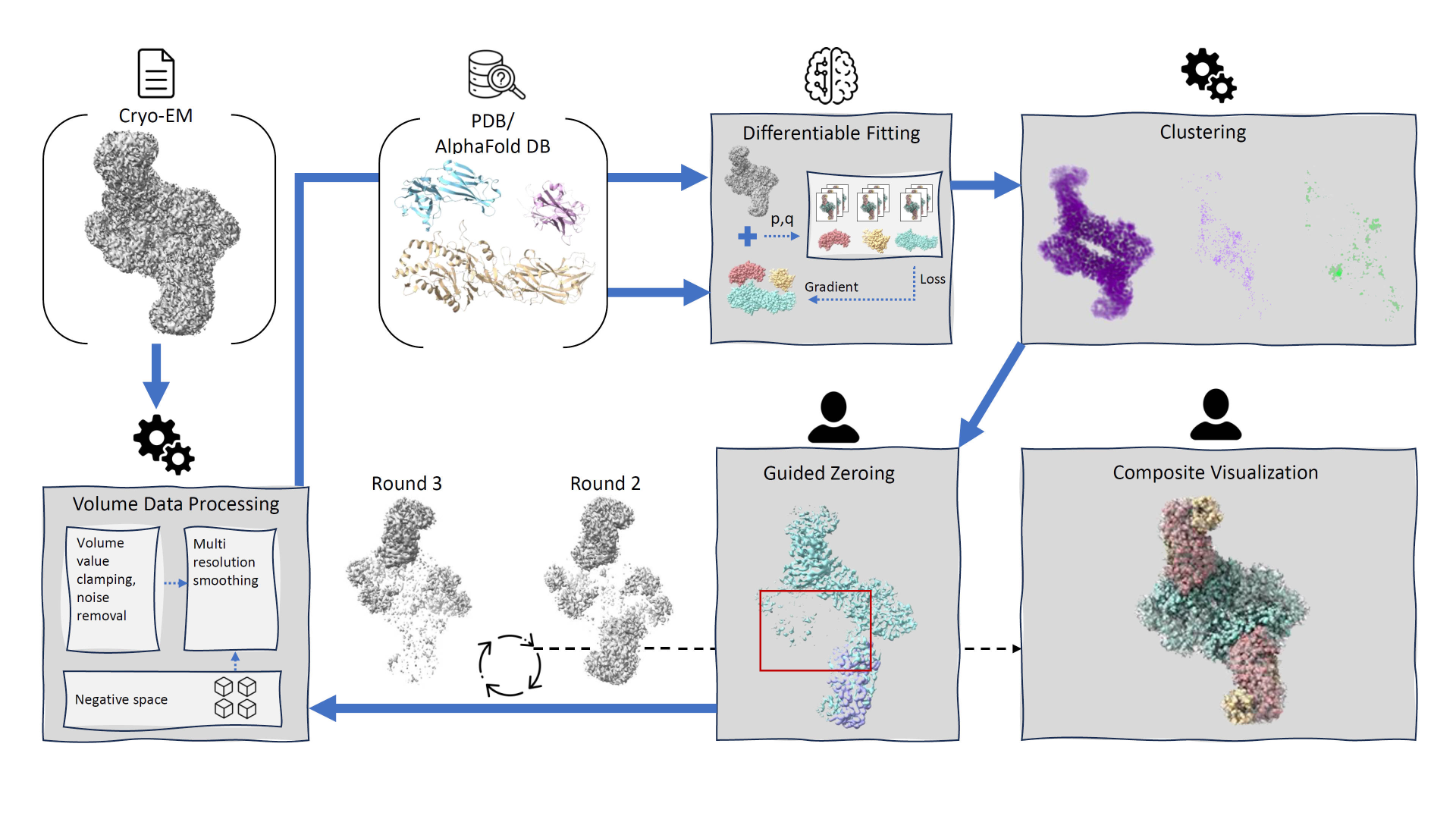
We introduce DiffFit, a differentiable algorithm for fitting protein atomistic structures into an experimental reconstructed Cryo-Electron Microscopy (cryo-EM) volume map. In structural biology, this process is necessary to semi-automatically composite large mesoscale models of complex protein assemblies and complete cellular structures that are based on measured cryo-EM data. The current approaches require manual fitting in three dimensions to start, resulting in approximately aligned structures followed by an automated fine-tuning of the alignment. The DiffFit approach enables domain scientists to fit new structures automatically and visualize the results for inspection and interactive revision. The fitting begins with differentiable three-dimensional (3D) rigid transformations of the protein atom coordinates followed by sampling the density values at the atom coordinates from the target cryo-EM volume. To ensure a meaningful correlation between the sampled densities and the protein structure, we proposed a novel loss function based on a multi-resolution volume-array approach and the exploitation of the negative space. This loss function serves as a critical metric for assessing the fitting quality, ensuring the fitting accuracy and an improved visualization of the results. We assessed the placement quality of DiffFit with several large, realistic datasets and found it to be superior to that of previous methods. We further evaluated our method in two use cases: automating the integration of known composite structures into larger protein complexes and facilitating the fitting of predicted protein domains into volume densities to aid researchers in identifying unknown proteins. We implemented our algorithm as an open-source plugin (github.com/nanovis/DiffFitViewer) in ChimeraX, a leading visualization software in the field. All supplemental materials are available at osf.io/5tx4q.